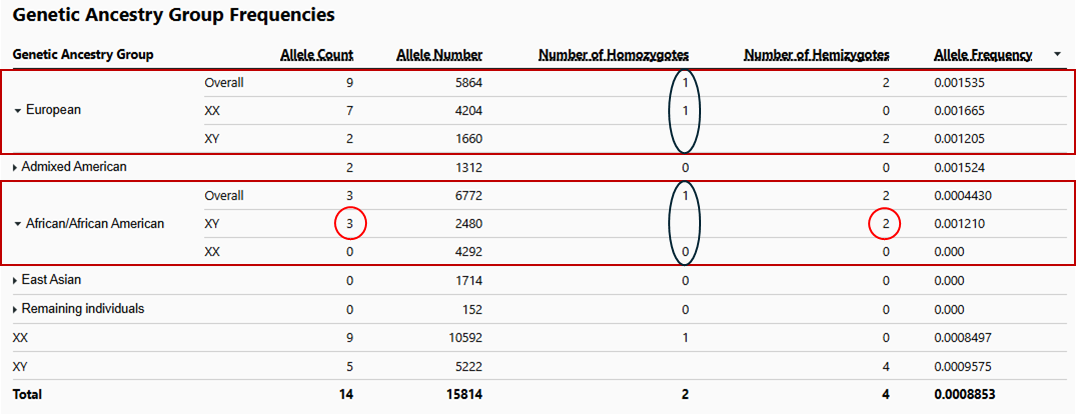
E.g. the number of males (XY) with the variant e.g. in the African subpopulation is ‘81’ (indicated as homozygotes), while the number of hemizygotes is ‘0’.
Is it intended to be this way, or is this a bug (which can be perhaps easily fixed)?
Had the same observation recently for some SNVs: tabix https://storage.googleapis.com/gcp-public-data--gnomad/release/4.1/vcf/exomes/gnomad.exomes.v4.1.sites.chrX.vcf.bgz chrX:284193-284193 | grep "\sT\sC\s" | tr ';' '\n' | grep '^AC_XY' AC_XY=531069
vs. gnomAD → 0
for me it turned out that the mismatch between hemizygous count on gnomad website and AC_XY field is coming from PAR regions… checking for the INFO flag non_par helps
We’ve just pushed a fix for the discrepancy for structural variants, those should now show the correct data. We’re continuing to look into the short-variant case.
I’ve checked a number of short variants outside of the PARs and it looks like the hemizygote count matches the XY count in each case. Thanks for bringing these to our attention.
Yes, it looks like the hemizygote number matches the XY number now in every cell (green highlights).
However, there seems to be one more discrepancy. There are some homozygotes displayed for males (which doesn’t make sense), and it also gets counted into the overall number of homozygotes (see yellow highlights). This only seems to be a problem for CNVs (and not for SNVs) on the X chromosome.
The issue was partially fixed (on Jun 27-28), but it remained partly erroneous. I asked again here to fix it, but nothing happened, so I’ve posted the remaining part of the error (i.e. on Sep. 3: GnomAD SVs v4, X-chromosome: homozygous males) as a new topic, to make sure it’s not getting considered to be already fixed. Can someone pls look into it?
Can you pls check out this CNV on the X-chromosome:
The number of homozygotes still seems to be erroneous, i.e. statistically highly unlikely (i.e. practically impossible) to expect this high number of homozygotes based on the reported allele counts (i.e. 158 total alleles, consisting of 63 homozygotes, 25 hemizygotes and 7 calculated heterozygotes). Therefore, based on the number of hemizygotes it is plausible that the reported number of ‘homozygotes’ in reality represents almost exclusively ‘heterozygous’ occurrences.
Sorry I missed your earlier message. I’ve just checked the original VCF from which this table was loaded, and the data displayed on the site does match what’s in that VCF. I’ll inquire with the relevant researchers to see if they can shed some light on this.
Thank you for your interest in gnomAD SV and CNV callset. I have looked into the duplication you mentioned, and here is what I found:
The variant is correctly represented in the browser according to the VCF.
You are correct that most female carriers of this duplication should be heterozygous rather than homozygous.
Upon tracing this event back through our SV discovery pipeline, I believe the overestimation of genotype is due to the presence of other large deletions (>100 Mb) on chromosome X in the same carriers, which overlap with this duplication. Our algorithm automatically increased the copy number of duplications under the assumption that overlapping deletions on the other allele could reduce the estimated copy number. These large overlapping deletions were later manually reviewed and confirmed to be false positives, and subsequently removed from the final VCF. However, the copy states of overlapping CNVs were not updated accordingly.
I greatly appreciate you bringing this issue to our attention, and we will work on updating the GATK-SV pipeline to prevent this in the future. In the meantime, I would like to point you to gnomAD v2 (gnomAD), where this duplication is reported with clearer frequencies and counts of homozygous versus heterozygous carriers.
Thank you again for highlighting this important point.
Thanks so much for looking into this. Let me share my thoughts by breaking down this question into 2 parts (i.e. male and female homozygous occurrences):
Female homozygotes: The presence of overlapping large deletions (>100 Mb) on chromosome X can certainly explain some of the perceived homozygous occurrences in females, if these large dels are frequent enough to co-occur with these CNVs on the other allele. Since you mentioned that these large dels were false positives which were eventually removed, will the gnomAD data be updated soon to eliminate these perceived homozygotes?
Male homozygotes: This phenomenon (i.e. overlapping large del on the other allele) doesn’t seem to explain the presence of homozygous occurrences in males (XY), since this gene (MAGT1) is not located to a pseudoautosomal region (PAR). The same is true for the above linked example in the DMD gene region (i.e. DMD is also not located to a PAR, while several homozygous occurrences are shown in males).
Lastly, I’ve checked the SV2.1 version earlier for this MAGT1 dup, and found some errors there as well, i.e. in the African subpopulation a homozygote is indicated which is not shown for females, and eventually it was counted as a hemizygote in the overall allele count:
To my experience, this problem seems to affect CNVs on the X chromosome, and it is probably more noticeable in the SVs v4 dataset (most likely because it has a larger allele count).
I think, if those entries are not factual errors in the data-tables on the gnomAD website (e.g. caused by a coding glitch), then another potential explanation could be (in addition to the overlapping large dels in females) if some of these copy gains would be triplications rather than dups. In general, would it be possible, when certain CNVs have some copy number variability which doesn’t reach your criteria to ascertain those as mCNVs (multiallelic CNVs), this might result in an edge case scenario (with weird looking duplication tables)?
You are correct regarding duplications: when they are not classified as multi-allelic CNVs, we assign a heterozygous genotype to samples with a copy number of 3, and a homozygous genotype to those with a copy number greater than 3. In some cases—though I would expect these to be rare—triplications may be assigned a homozygous genotype, as we are not able to phase the two alleles using short-read WGS data. With the availability of long-read sequencing in the future, this limitation should no longer be an issue.
I just wanted to add an interesting paper to this discussion thread.
An apparent homozygous duplication in gnomAD SVs v4.1.0 in the BRCA1 gene (DUP_CHR17_71E8B388, hg38 17:43120665-43127972, size: 7,307 bp) is actually (likely) a tandem triplication, as it was reported by Caputo 2021 (PMID: 34202044). The authors of the study found this variant in multiple families and confirmed the tandem triplication of exons 1 & 2 of BRCA1 by molecular combing. They reported the triplication variant with almost identical breakpoints (hg38 17:43120714-43127972) and similar size (7,259 bp).
This suggests that for duplication CNVs when the number of apparent homozygotes in gnomAD is larger than the expected number of homozygotes, they are likely to represent triplication variants.